








This 90-minute training session is about how QIAGEN Ingenuity Pathway Analysis (IPA) allows visualization of molecular intricacy and variations at multiple levels such as transcriptome, proteome, and metabolome. Through a series of brief technical vignettes, it is demonstrated how to:
· Generate associations among molecular signatures obtained via integrating multi-omics data
· Extract mechanisms from multi-omics data for precision medicine
· Disease stratification based on multi-omics profiles
· Map disease networks among targets and indications
Transcript discovery using RNA-seq is an important step in refining eukaryotic genome annotation by complementing in-silico gene predictions with experimental evidence for genes, transcripts, splice variants, and their expression levels. Best practice is to include evidence from both long- and short-read NGS data, which reduces challenges stemming from inference of transcripts and their splice variants from partial observations or rarely used splice junctions resulting in read-through events.
The new Transcript Discovery Plugin for CLC Genomics Workbench and the CLC Genomics Server allows joint analysis of both long PacBio and short Illumina reads from RNA-seq libraries. The plugin works by mapping RNA-Seq sequencing reads to a genomic reference while permitting large-gap alignments (to account for introns), followed by a transcript discovery process where transcripts and genes are inferred from the read mappings. If genome annotations already exist for the genome at hand, these can be updated based on the experimental evidence from the RNA-seq data, thus generating new transcript and gene annotation tracks.
Central to the Transcript Discovery Plugin is the Large Gap Read Mapping step, which models the presence of introns in the genome reference sequence, but are expected to be absent from the corresponding transcriptome. RNAseq reads are iteratively mapped to the reference genome using the CLC Genomics Workbench read mapper. The Transcript Discovery step then predicts genes, transcripts, and coding regions (CDS) from the read mapping results, while considering prior knowledge on gene and transcript models and the coding potential of predictions. The estimation is also aided by several filters applied to the mapped reads, such that coverage, splice evidence and splice signatures are considered, while duplicated, non-specific matching, read-through and chimeric reads are disregarded.
Rich tables of predicted features with columns describing their various evidences can be viewed as genome tracks and inspected together with the read mappings. Furthermore, examination of the Rejected Events table can lead to insights on filtering settings that can be fine-tuned until sensitivity and specificity are in balance. An example of some of the output from this plugin can be seen in Figure 1.
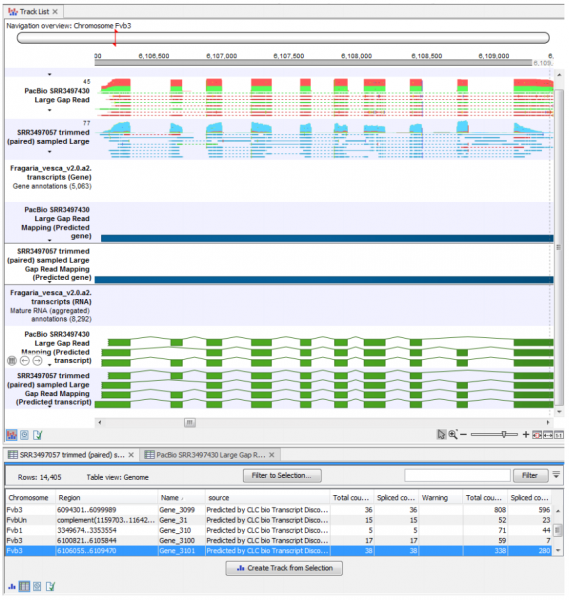
Figure 1: RNA-seq mapping and annotation tracks identified using the Transcript Discovery tool seen in a Track List, with the Predicted Gene annotations opened as a table in split view.
The Transcript Discovery Plugin for the CLC Genomics Workbench comes with a fully documented manual and a quick-start tutorial. The manual both documents the use of the plugin, and contains tips on how to obtain the best results by a) optimizing the sequence in which iterative analysis steps are performed and b) conducting combined versus sequential processing of multiple replicates and tissues of origin.
The quick-start tutorial provides a step-by-step guide to performing RNA transcript discovery from your RNA-seq libraries. Part of a recently published dataset (1) containing both PacBio and Illumina reads from the Wild Strawberry Fragaria vesca is provided in the tutorial, which can be used to re-annotated the reference genome. In their paper, Li et al., 2018 annotated approximately 33,000 genes using the full dataset (available from SRA). To enable users to walk through the tutorial on a broad-range of computers, we provide a link to a smaller subset of the reads. Nonetheless, these still are enough to annotated over 14,000 genes, including several new genes and transcripts that were not presented in the original paper.
The Transcript Discovery Plugin is a free add-on for the CLC Genomics Workbench that provides an easy-to-use tool-set for evidence-based RNA-seq annotation of genomes. The plugin can be downloaded and installed directly from within the Plugin Manager CLC Genomics Workbench. Alternatively, an installer file can be download from our Workbench Plugins page, where a CLC Genomics Server version of the plugin is also available.
In the past several months, customers have performed remarkable work with CLC Genomics Workbench, a tool that offers customizable bioinformatics solutions for genomics, transcriptomics, epigenomics, and metagenomics. Many scientists rely on this tool for genome assembly and interpretation every day and here, we look at a handful of recent publications that have been supported by CLC Genomics Workbench.
De novo transcriptome assembly and comprehensive expression profiling in Crocus sativus to gain insights into apocarotenoid biosynthesis
First author: Mukesh Jain
This paper in Nature’s Scientific Reports journal comes from scientists in New Delhi who conducted transcriptome sequencing to better understand important biological mechanisms in saffron. They identified transcription factors, differentially expressed genes, and more. The team used CLC Genomics Workbench in the transcriptome assembly and the analysis of differentially expressed genes.
Complete coding sequence of Zika virus from Martinique outbreak in 2015
First author: G. Piorkowski
Scientists in France studied a strain of the Zika virus isolated from a patient in the Caribbean. In this publication, they report the full coding sequence of the virus, an essential resource for the ongoing battle against Zika virus. They used CLC Genomics Workbench to analyze the data generated with an Ion Torrent sequencer.
Effective de novo assembly of fish genome using haploid larvae
First author: Yuki Iwasaki
Published in the journal Gene, this marine genomics study from scientists in Japan demonstrates the utility of sequencing haploid fish larvae — in this case, yellowtail fish — to obtain a diploid genome assembly. They used the Proton sequencer and compared the performance of overlap-layout-consensus (OLC) and de Bruijn graph (DBG) assemblers. CLC Genomics Workbench, which uses a DBG approach, outperformed another tool in the study.
Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line
First author: Reed S. Shabman
This study, an editor’s pick in the mSphere journal, comes from scientists at the J. Craig Venter Institute and other organizations who were analyzing the transcriptome of a microbat when they discovered RNA from a previously unidentified herpesvirus. They used CLC Genomics Workbench for assembly and finishing of the viral genome.
Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae
First author: Kai Zhou
In this Scientific Reports paper, scientists from China and the Netherlands tracked the transmission path of a Klebsiella pneumoniae strain using genome-based phylogenetic analysis. They used a mapping unit in CLC Genomics Workbench to find genes associated with antibiotic resistance and pathogen virulence, and detected SNPs by mapping isolate sequence data with the same tool.
Read more about CLC Genomics Workbench



