








You asked for it by popular demand, and we’re here to deliver. In this training, we’ll cover how antimicrobial resistance (AMR) is used for isolation or can be easily integrated with other functionalities such as multi-locus sequence typing (MLST).
In this training, we’ll cover:
• Introduction to AMR, MLST and relevant databases
• QIAGEN Microbial Insight - Antimicrobial Resistance database (QMI-AR)
• Importing data and downloading needed databases
• Finding resistance with nucleotide DB, PointFinder and ShortBRED
• Integrating AMR with MLST
• Exporting high-resolution graphics and result tables
In this 90-minute training, you will learn how to easily analyze wastewater samples to detect pathogens (SARS-COV2, etc.) using QIAGEN CLC Genomics Workbench software.
You will learn how to:
• Importing reads
• Open and modify prebuilt workflow (analysis pipeline)
• Install and execute workflow
• Review QC reports
• Perform genome visualization
• Export the consensus sequence in FASTA format to upload to Pangolin
• Create a SNP tree of the consensus sequence and overlay Pangolin information
• Export graphical and tabular results
QIAGEN CLC Genomics Workbench provides tools and workflows for a broad range of bioinformatics applications, including microbiome analysis, isolate characterization through SNP and K-mer trees using NGS data, and antimicrobial resistance characterization. CLC Genomics Workbench is widely used for analyses of bacterial, viral and eukaryotic (fungal) genomes and metagenomes.
Topics covered in this webinar include:
I. Overview of different tools within MGM application and research areas supported
II. Focused review of isolate typing and characterization
a. Importing data
b. Utilization of metadata
c. Downloading and managing references
i. Database of Isolates/ Resistances/ MLST
d. Walk through of Type a Known Species workflow
i. Review details for each Isolate
e. Creating SNP profiles to specific reference
f. Generate a SNP tree for isolate comparison
g. Export tabular and high-quality graphical outputs in wide range of file formats
Click here to register.
Microbes play essential roles in human, animal and plant health. Knowing the composition and diversity of a microbial community is the first step in understanding microbe-microbe and microbe-host interactions, revealing key players in health and disease and discovering major modulators. Next-generation sequencing in microbiome research has accelerated data generation and changed our perception of the complexity and structure of microbial communities. However, microbiome data sets are typically high-dimensional, adding further complexity to data analysis and processing.

In our white paper, “Characterizing the Microbiome through Targeted Sequencing of Bacterial 16S rRNA and Fungal ITS Regions,” we demonstrate the power of the CLC Microbial Genomics Module for investigating complex microbial communities.
We used the original data of Purahong et al.’s work from the April 2016 edition of Molecular Ecology “Life in the leaf litter: novel insights into community dynamics of bacteria and fungi during litter decomposition,” to analyse amplicon sequencing data for profiling microbial communities via clustering reads into operational taxonomic units. Purahong’s study explored the dynamic interplay of bacteria and fungi during leaf degradation over a one-year period in response to fluctuations in nutrient availability.
We demonstrate how the usage of our pre-configured CLC workflows can ease analysis on-boarding, reduce hands-on time, and ensure consistency and reproducibility in microbiome analyses.
Purahong W, et al. (2016). Life in the leaf litter: novel insights into community dynamics of bacteria and fungi during litter decomposition. Molecular Ecology 25, 4059.
Microbial communities contribute more than half of all the cells our bodies are composed of. And not surprisingly, the taxonomic and genetic makeup of microbiomes is closely linked to the health of humans, animals and plants.
Yet especially the functional genetic composition of microbiomes is hard to establish and current metagenomics tools struggle with correctly predicting functional composition or changes in function between microbiome samples [Lindgreen et al. 2015].
What if you could access tools to de novo assemble metagenome data, reliably predict functional elements, and identify statistically significant changes in function between samples? And what if these tools were fully integrated into the industry standard for scientist-friendly NGS data analysis, and came along with a toolbox that has been optimized for microbiologists?
CLC Genomics Workbench, CLC Microbial Genomics Module and the MetaGeneMark plugin deliver superior performance, a fully integrated user experience and come bundled at a competitive price.
Accuracy of results
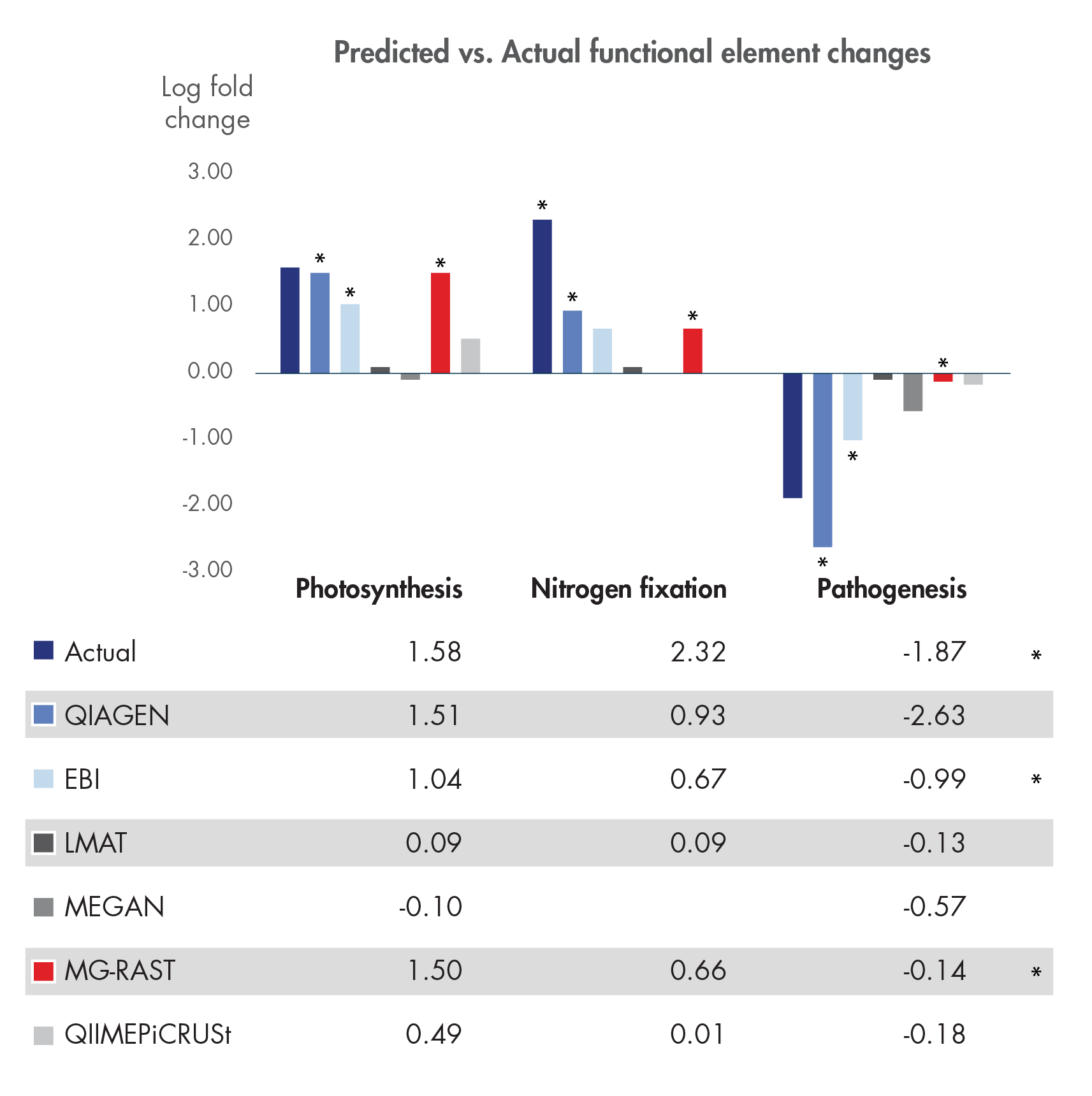
Figure 1. Assigning and tracking gene function in metagenomes with confidence.
Lindgreen et al. published a comprehensive, independent evaluation of 14 different whole metagenome analysis toolkits in Nature Scientific Reports in January 2016. We here compare our solution to the sole five toolkits out of the fourteen that allow functional metagenome analysis using the test data published by Lindgreen et al. Statistical comparison (Edge test performed in CLC Genomics Workbench) of pairwise differential abundance of the individual functional elements predicted in the two test communities detects a statistically significant difference for all of the three functional elements that were analyzed in the paper: photosynthesis, nitrogen fixation and pathogenesis (all p-values < 0.01). Fold-changes predicted using our tools capture the expected overall pattern of functional changes and estimate the actual fold-change with higher precision than any other tool in all three functional roles.
* indicates tools that consistently predicted changes correctly with statistically significance.
Detecting gene function in microbial communities based on metagenomic data is hard. Correctly measuring changes in the functional makeup between different metagenome samples is even harder.
Lindgreen et al. showed that most of the benchmarked open source tools failed to correctly predict such changes at levels that are statistically significant.
With our solution for microbial genomics you can more accurately detect and quantify functional elements in a sample. And the included statistical tools allow you to confidently measure statistically significant changes in function between samples.
Multi-sample comparison is used to detect functional changes between samples and to identify samples with similar or diverging functional genomic elements. Data can be grouped and analyzed in the context of your sample-metadata.
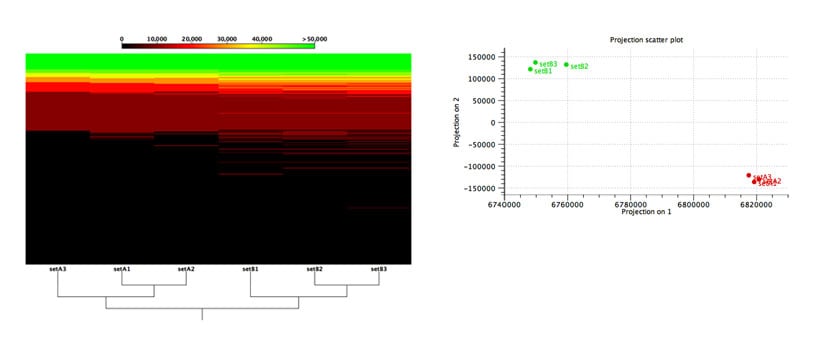
Figure 2: Functional comparison across microbiome samples.
Evaluation of an algorithm’s capabilities in detecting functional changes in metagenomes is notoriously hard because the ground truth is unknown and there exist no gold-standard datasets. To overcome these difficulties, Stinus Lindgreen et al. created six datasets from two synthetic microbial communities for his benchmarks: three (A1, A2 and A3) from the A community and three from the B community. To control the functional content, he created the two communities, A and B, with a selected set of species with known functional capabilities: Cyanobacteria (photosynthesis), Bradyrhizobium (nitrogen fixation) and Rhizobium (nitrogen fixation) were more abundant in community A, while a set of known pathogens where more abundant in community B.
As shown in Figure 2, our tools were able to reliably separate samples from the two different communities based on the relative abundance of their predicted functional content.
Our accurate assignment of gene function depends on a novel metagenome assembler producing higher quality assemblies compared to leading alternatives. Table 1 illustrates how our metagenome assembler compares favorably when it comes to misassemblies, InDels, mismatch errors, and other quality metrics.
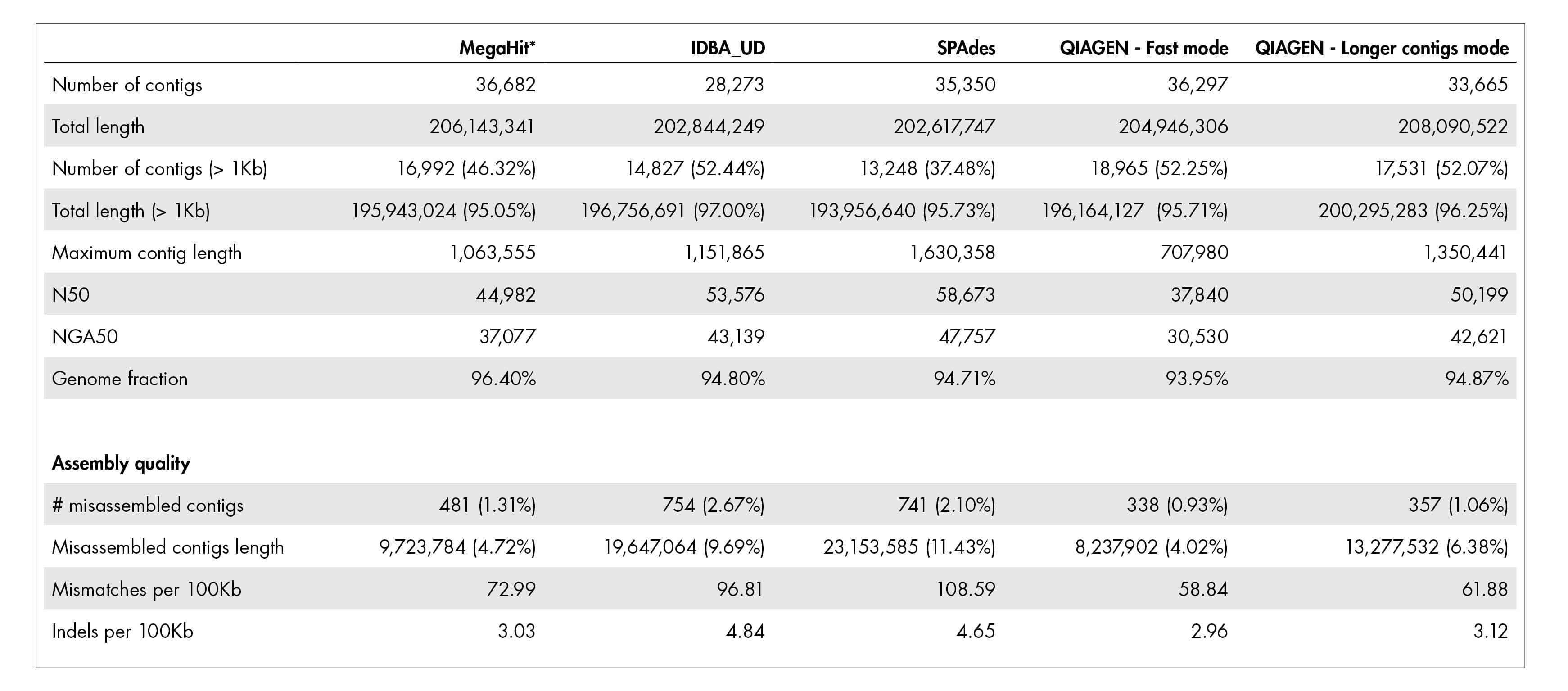
Table 1: Quality of metagenome assembly.
The QIAGEN metagenome assembler delivers superior assembly quality resulting in more accurate annotation of functional genetic content. A dataset published by Shakya et al. 2013 was used for this benchmark. The actual number for “Total length” and “Total length (>1kb)” should be close to 209,845,413 bases.
Run time and compute resource requirements are important when sample volume is high.
We have benchmarked the metagenome assembler included in our microbial genomics solution against leading metagenome assemblers using a dataset by Shakya et al. 2013. Shorter run time and greater compute resource efficiency was consistently demonstrated compared to other leading assemblers.
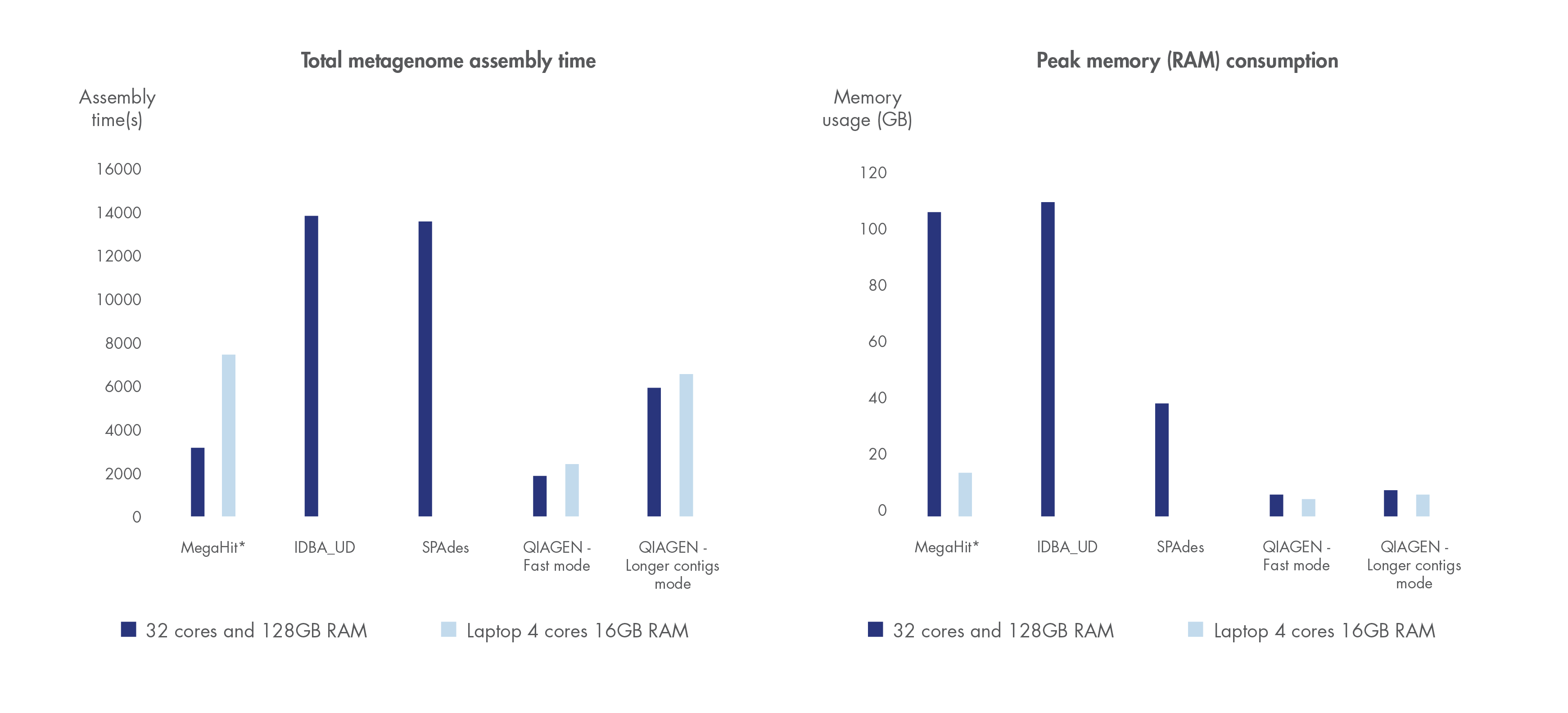
Figure 3. Best in class metagenome assembly.
Accelerated algorithms result in metagenome assembly that outcompetes leading alternatives in run time and compute resource consumption. *Note that MegaHit is able to scale its memory consumption down by sacrificing run time.
Increase walk away time
To increase walk away time, users can use the Workflow feature in CLC Genomics Workbench to combine the analysis steps 2 through 7 listed below into a preconfigured one-click workflow. Workflows are capable of batch processing many samples increasing walk-away time.
Analysis steps in functional metagenomics workflow:
1 → Import of multiple whole metagenome sample read datasets and association of metadata to each sample.
2 → QC and trimming of whole metagenome reads.
3 → De novo assembly of each sample read dataset into high-quality contigs using the new De Novo Assemble Metagenome tool.
4 → Locate coding sequences (CDS) in the resulting contigs using the third-party MetaGeneMark genefinder plugin for the CLC workbenches.
5 → Annotate CDSs with Gene Ontology (GO) terms and Pfam protein families or Best BLAST Hits using one of the two new tools, Annotate CDS with Pfam or Annotate CDS with Best BLAST Hit, respectively.
6 → Map the input reads back to the annotated contigs using the built-in Map Reads to Reference tool in the CLC workbenches.
7 → Build a functional abundance profile of each sample using the Build Functional Profile tool
8 → Merge the functional abundance profiles for all samples into one profile using the Merge Functional Profile tool.
9 → Visualize the individual and merged functional abundance profiles, perform filtering based on abundance, and apply the different options for showing the abundance profiles in the context of metadata.
10→ Perform hierarchical clustering and statistical analysis based on the relative abundance of functional elements in the samples.
Find out more about our microbial genomics solution
Assembling gold standard reference genomes for microbial pathogens has become fast and easy. We're proud to bring you a new feature allowing rapid error correction and assembly of PacBio data.
The latest version of CLC Genome Finishing Module introduces tools for error-correction and de novo assembly of raw PacBio reads. High quality assemblies can be generated in a fraction of the time that is needed by leading alternatives.
CLC Genome Finishing Module consumes less than 10 percent of the memory used by alternative solutions, while completing the assembly faster. The Module runs on all major operating systems, and fully integrates into the widely used CLC Genomics Workbench and CLC Genomics Server solutions. The included preconfigured workflow makes it extremely user-friendly.
Benchmarking
We compared the performance of the industry standard HGAP1 when run on a high performance computer to the performance of the workflow "PacBio De Novo Assembly Pipeline" included in CLC Genome Finishing Module. Please note that our PacBio De Novo Assembly Pipeline was run on a standard laptop for this comparison.
1Chin et al. (2013), Nature Methods, 10(6), 563–569
Not convinced yet? Try it out

We are pleased to announce the launch of CLC Microbial Genomics Module, a powerful new tool that enables researchers focused on sequencing and characterizing bacterial communities, for example in food production, agricultural research and infectious disease to visually explore and analyze microbiomes.
With this new module, researchers can now investigate microbial communities as biomarkers that are indicative of patient health, changing yields of agricultural crops or livestock, or of the emergence of public health threats.
For information on the CLC Microbial Genomics Module, please visit https://digitalinsights.qiagen.com/plugins/clc-microbial-genomics-module/ or read the official press release below.
Press Release
QIAGEN launches new bioinformatics solution for microbial genomics
REDWOOD CITY, California, June 15, 2015 – QIAGEN Bioinformatics, today announced the launch of its CLC Microbial Genomics Module which enables academic and commercial researchers focused on food production, agbio and infectious disease to visually explore and analyze microbiomes. This new add-on module is available via an integration with the widely used CLC Genomics Workbench and CLC Genomics Server software solutions.
“As an emerging field, the study of microbiome genomics suffers from a lack of integrated solutions that allow us to seamlessly go from raw next-generation sequencing data to the statistical analysis of results and metadata to the interactive visualizations that provide the insights,” said Gautam Dantas, PhD, Associate Professor at the Washington University School of Medicine. “The CLC Microbial Genomics Module directly addresses this challenge by offering an end-to-end workflow that integrates the algorithms, interactive visualization and powerful statistical tools we need into a single solution.”
The CLC Microbial Genomics Module is a comprehensive solution for microbiome profiling that provides preconfigured automatic workflows and complete analyses for individual samples or entire experiments. The software’s tools do not require bioinformatics expertise and enable visual microbiome composition browsing and analysis at different taxonomic levels and in the context of metadata. Operational Taxonomic Units (OTU)-clustering and taxonomic annotation are based on common reference databases such as Greengenes, Silva and UNITE. Principal Coordinate Analysis (PCoA) results can be explored three dimensionally in the context of sample metadata, to understand which samples share similar features.
“NGS technology is expanding with an ever increasing variety of applications, and in few fields are samples and data more complex than the intricate world of microbial communities and their interplay with their hosts and the environment,” said Dr. Arne Materna, global product manager at QIAGEN Bioinformatics. “CLC Microbial Genomics Module further expands our portfolio for researchers utilizing next-generation sequencing data. With this new module, researchers can now investigate microbial communities as biomarkers that are indicative of patient health, changing yields of agricultural crops or livestock, or of the emergence of public health threats.”
Bioinformatics is a key growth driver for QIAGEN. The Company’s integration of Ingenuity Systems, CLC bio and BIOBASE has created the industry-leading provider of integrated bioinformatics solutions and expertly curated content (such as Ingenuity Knowledge Base and HGMD®) for the analysis, interpretation and reporting of biological data. For information on the CLC Microbial Genomics Module, please visit https://digitalinsights.qiagen.com/plugins/clc-microbial-genomics-module/.
About QIAGEN
QIAGEN N.V., a Netherlands-based holding company, is the leading global provider of Sample to Insight solutions to transform biological materials into valuable molecular insights. QIAGEN sample technologies isolate and process DNA, RNA and proteins from blood, tissue and other materials. Assay technologies make these biomolecules visible and ready for analysis. Bioinformatics software and knowledge bases interpret data to report relevant, actionable insights. Automation solutions tie these together in seamless and cost-effective molecular testing workflows. QIAGEN provides these workflows to more than 500,000 customers around the world in Molecular Diagnostics (human healthcare), Applied Testing (forensics, veterinary testing and food safety), Pharma (pharmaceutical and biotechnology companies) and Academia (life sciences research). As of March 31, 2015, QIAGEN employed approximately 4,300 people in over 35 locations worldwide. Further information can be found at www.qiagen.com.



