








When it comes to taxonomic profiling of the microbiome, amplicon sequence variants (ASVs) is a method that has unique benefits compared to the traditional method of sequence similarity-based operational taxonomic unit (OTU) clustering. This training’ll cover how to effectively do taxonomic profiling using QIAGEN CLC Genomics Workbench’s ASV tools and a customizable workflow.
Specifically, you’ll learn how to:
1. Collect and bin sequence variants for each sample
2. Merge AVS tables to compare profiles
3. Assign the taxonomic profile/organism
4. Export high-resolution graphical and comprehensive tabular results
If you’re new to QIAGEN CLC Genomics Workbench and are interested in a trial, learn more here:
https://qiagen.showpad.com/share/I6MqdhIybpllyvuYW3AKy
QIAGEN CLC Microbial Genomics Module provides tools and workflows for many various bioinformatics applications, including microbiome analysis, isolate characterization, functional metagenomics and antimicrobial resistance characterization. The module supports the analysis of bacterial, viral and eukaryotic (fungal) genomes and metagenomes.
This training focuses on amplicon-based taxonomic profiling (16S/18S/ITS sequencing OTU clustering). Specifically, we’ll cover together:
• An overview of the different tools within QIAGEN CLC Microbial Genomics Module and the research areas supported
• Taxonomic profiling
• Importing data
• Utilization of metadata
• Downloading and managing references
• The OTU clustering workflow (analytical pipeline)
• Downstream processing of abundance tables
• Creating and exporting high-quality graphics
In our recent white paper we describe how to investigate the functional potential of a microbial community in a polar desert in Antarctica using metagenomic shotgun sequencing data. In the original paper (1), the authors supplemented their microbiome data with qPCR analyses to investigate the expression of the most interesting genes discovered in the functional profiles to support their hypothesis that the microbial community survive by scavenging atmospheric trace gases. However, what if they had instead included RNA-seq transcriptomic data to evaluate gene activity in their samples? In this post, we show you how to add transcriptomics data to a microbiome survey using the tools of CLC Genomics Workbench.

The example below presents a de novo assembly based approach to metatranscriptomic analysis using CLC Genomics Workbench and the Microbial Genomics Module. There are, in fact, multiple approaches to performing metatranscriptomics data analysis, depending on the specific questions you may have. For a deeper review on best-practices in metatranscriptomics analysis we recommend you review Bashiardes et. al. (2), or read published examples where CLC Genomics Workbench was used for metatranscriptomics research; some recent interesting examples include a study of thehoney bee (3) and termite (4) microbiomes and their associated metatranscriptomes.
The example metatranscriptomic pipeline presented below consists of two parts (shown in Figure 1). Part 1 includes: assembling the metagenome; grouping contigs into bins to reconstruct the microbial genomes; and finding and annotating genes. It is also described in further detail in our recent white paper on Antarctic microbiome profiling. A common approach and caveat of comparing metatranscriptomes from multiple samples is often to create a “co-assembly” across your samples that serves as a single reference list of contigs and genes for the downstream RNAseq analysis. A good example of this approach can be found in Marynowska et. al. (4).
Part 2 of the analysis pipeline involves adding the transcriptomic data to supplement the metagenomic survey with information on gene activity. Part 2 is the focus of this post and will be described below.
CLC Genomics Workbench include a suite a of tools designed for analyzing gene expression data. For this blog post, we will use just only a few of them. The RNA-Seq Analysis tool will start with mapping reads to the genome and the coding sequences. The tool requires a file with the reference genome and a file with annotations for protein coding sequences (CDS) or genes. If these are not already available from Part 1 of the pipeline (Figure 1), they can be generated using Track Tools -> Track Conversion -> Convert to Tracks. This will take an annotated genome or list of contigs as input and generate individual track files. Additional details on this conversion step can be found in our manual. In this case we need to generate a track for the genome and one for the annotated coding regions. From the read mappings, reads are categorized and assigned, and expression values are calculated. The output from the RNA-Seq Analysis tool is a table describing for each gene the number of reads mapped, the number of reads per kilobase gene, and the expression value. The results can be visualized in a track list along with the genes and the read mappings (Figure 2). The track list is interactively linked to the results table, and marking a CDS of interest in the table view, will shift the focus of the track list to that particular region.

From the track view read mappings can be manually inspected by zooming in on individual genes (Figure 3). In the case of the desert soil microbiome in Antarctica, genes supporting the use of atmospheric trace gases as carbon and energy sources could be searched out from the table, and the expression values inspected.

If your microbiome investigation involves comparing microbial communities at different times or under different conditions, transcriptomes can be compared across multiple states. This analysis can be performed with the tool Differential Expression for RNA-Seq. The tool performs a statistical test of the differential expression of two or more samples. The output is a table displaying for each gene, the fold change and the p-value for the statistical comparison. From this list, genes significantly changing expression levels under different biological conditions can be found.
CLC Genomics Workbench contain several additional tools for analyzing RNA-Seq data for more sophisticated comparisons and visualizations than what have been shown here. If you are interested in learning more or trying out the functionalities, you can always download a free trial.
We recently attended the 11th annual Sequencing, Finishing and Analysis in the Future (SFAF) meeting, hosted by Los Alamos National Laboratory and held in Santa Fe, NM, also known as “The City Different.” With an emphasis on genome sequencing, assembly, and applications of NGS, it was an action-packed three days of meetings, demos, poster presentations, and networking.
On the afternoon before SFAF started, QIAGEN Bioinformatics hosted a successful workshop for 30 attendees, which provided hands-on training on our Microbial Genomics Pro Suite. This user-friendly, scalable solution includes CLC Genomics Workbench and a series of plugins, enabling microbiologists to fully optimize the microbial genomics and metagenomics analytical workflow. Our session included an introduction to the capabilities of the solution, including metagenomics workflows, pathogen typing, and outbreak analysis. Resources for this training are available on our website and include an explanatory video, entitled “Introduction to Metadata Handling for CLC Microbial Genomics Module.”
SFAF 2016 was a wonderful event, bringing us the chance to strengthen existing relationships and meet researchers who are using our solutions on fascinating projects. It also brought us the opportunity to discuss benchmarks for our microbial genomics solution. We are looking forward to following up with our new contacts — some of whose work we hope to feature on this blog and share with you in the not-too-distant future.
Learn More about our Microbial Genomics Solution
We recently had a fantastic week in Boston, where top educational institutions meet innovative life sciences. The Boston Convention Center, located in the buzzing Seaport District, was host to the ASM Microbe 2016.
The agenda was packed with symposia, workshops, and poster sessions, there were blue skies outside the convention center, and hundreds of conference attendees visited our booth to discuss microbiology from sample to insight. We were delighted to see strong interest in our bioinformatics solutions - particularly how CLC Genomics Workbench, our new QIAGEN CLC Genomics ProSuite, and Ingenuity Pathway Analysis help turn data into insights.
This year we highlighted how our solutions are being used to investigate pathogen genomes and outbreaks, to profile microbiomes, and to study host response to infection. Our new pipeline for functional metagenomics drew significant interest, due in part to Dr. Rita R. Colwell's fascinating presentation on the importance of rapid taxonomic and functional analysis of microbiome data in the case of enteric infections. You can watch the presentation below.
To give QIAGEN CLC Genomics ProSuite a try, download your trial today!
Learn More about our Microbial Genomics Solution
Clinical Metagenomics for Rapid Detection of Enteric Pathogens and Characterization of the Intestinal Microbiome in Health and Disease
Presented by Rita R. Colwell
ASM Microbe 2016 is the largest gathering of microbiologists and we'll of course be there to present our microbial genomics solution. The meeting is held June 16–20, 2016 in Boston and showcases the best microbial sciences in the world.
You can find us at booth #1822, where you can stop by for a chat and a demonstration of our solutions. We'll also be hosting a workshop and two poster presentations.
Clinical Metagenomics for Rapid Detection of Enteric Pathogens and Characterization of the Intestinal Microbiome in Health and Disease
Speaker: Dr. Rita Colwell, CosmosID Inc.; Center for Bioinformatics and Computational Biology & Maryland Pathogen Research Institute, University of Maryland; Bloomberg School of Public Health, Johns Hopkins University
High-throughput sequencing, combined with high-resolution metagenomic analysis, provides a powerful diagnostic tool for clinical management of enteric disease. 45 patient samples of known and unknown disease etiology and 20 samples from health individuals were subjected to next-generation sequencing. Subsequent metagenomic analysis identified all microorganisms (bacteria, viruses, fungi, and parasites) in the samples, including the expected pathogens in the samples of known etiology. Multiple pathogens were detected in the individual samples, providing evidence for polymicrobial infection. Patients were clearly differentiated from healthy individuals based on microorganism abundance and diversity. The speed, accuracy, and actionable features of the CosmosID MetaGenID bioinformatics and curated GenBook® databases, implemented in the QIAGEN CLC Genomics ProSuite, and the functional analysis, leveraging the QIAGEN functional metagenomics workflow, provide a powerful tool contributing to the revolution in clinical diagnostics, prophylactics, and therapeutics that is now in progress globally.
Date: Sunday, June 19 at 12:45 p.m. - 1:30 p.m.
Location: Industry & Science Showcase A – Booth 144
Bioinformatics Workflow For Metagenomics Data Analysis
Presenter: Marta Matvienko
Presentation time: Friday, June 17, 2016 at 12:30 p.m. – 2:30 p.m.
Poster board number: FRIDAY-667
Functional Metagenomics, A Novel Qiagen Bioinformatics Pipeline Enables Fast High-Quality Analysis Of The Functional Content Of Microbial Communities
Presenter: Andreas Sand
Presentation time: Friday, June 17, 2016 at 12:30 p.m. – 2:30 p.m.
Poster board number: FRIDAY-038
We're looking forward to seeing you in Boston!
More information about CLC Microbial Genomics Module
In the past several months, customers have performed remarkable work with CLC Genomics Workbench, a tool that offers customizable bioinformatics solutions for genomics, transcriptomics, epigenomics, and metagenomics. Many scientists rely on this tool for genome assembly and interpretation every day and here, we look at a handful of recent publications that have been supported by CLC Genomics Workbench.
De novo transcriptome assembly and comprehensive expression profiling in Crocus sativus to gain insights into apocarotenoid biosynthesis
First author: Mukesh Jain
This paper in Nature’s Scientific Reports journal comes from scientists in New Delhi who conducted transcriptome sequencing to better understand important biological mechanisms in saffron. They identified transcription factors, differentially expressed genes, and more. The team used CLC Genomics Workbench in the transcriptome assembly and the analysis of differentially expressed genes.
Complete coding sequence of Zika virus from Martinique outbreak in 2015
First author: G. Piorkowski
Scientists in France studied a strain of the Zika virus isolated from a patient in the Caribbean. In this publication, they report the full coding sequence of the virus, an essential resource for the ongoing battle against Zika virus. They used CLC Genomics Workbench to analyze the data generated with an Ion Torrent sequencer.
Effective de novo assembly of fish genome using haploid larvae
First author: Yuki Iwasaki
Published in the journal Gene, this marine genomics study from scientists in Japan demonstrates the utility of sequencing haploid fish larvae — in this case, yellowtail fish — to obtain a diploid genome assembly. They used the Proton sequencer and compared the performance of overlap-layout-consensus (OLC) and de Bruijn graph (DBG) assemblers. CLC Genomics Workbench, which uses a DBG approach, outperformed another tool in the study.
Isolation and Characterization of a Novel Gammaherpesvirus from a Microbat Cell Line
First author: Reed S. Shabman
This study, an editor’s pick in the mSphere journal, comes from scientists at the J. Craig Venter Institute and other organizations who were analyzing the transcriptome of a microbat when they discovered RNA from a previously unidentified herpesvirus. They used CLC Genomics Workbench for assembly and finishing of the viral genome.
Use of whole-genome sequencing to trace, control and characterize the regional expansion of extended-spectrum β-lactamase producing ST15 Klebsiella pneumoniae
First author: Kai Zhou
In this Scientific Reports paper, scientists from China and the Netherlands tracked the transmission path of a Klebsiella pneumoniae strain using genome-based phylogenetic analysis. They used a mapping unit in CLC Genomics Workbench to find genes associated with antibiotic resistance and pathogen virulence, and detected SNPs by mapping isolate sequence data with the same tool.
Read more about CLC Genomics Workbench
Microbial communities contribute more than half of all the cells our bodies are composed of. And not surprisingly, the taxonomic and genetic makeup of microbiomes is closely linked to the health of humans, animals and plants.
Yet especially the functional genetic composition of microbiomes is hard to establish and current metagenomics tools struggle with correctly predicting functional composition or changes in function between microbiome samples [Lindgreen et al. 2015].
What if you could access tools to de novo assemble metagenome data, reliably predict functional elements, and identify statistically significant changes in function between samples? And what if these tools were fully integrated into the industry standard for scientist-friendly NGS data analysis, and came along with a toolbox that has been optimized for microbiologists?
CLC Genomics Workbench, CLC Microbial Genomics Module and the MetaGeneMark plugin deliver superior performance, a fully integrated user experience and come bundled at a competitive price.
Accuracy of results
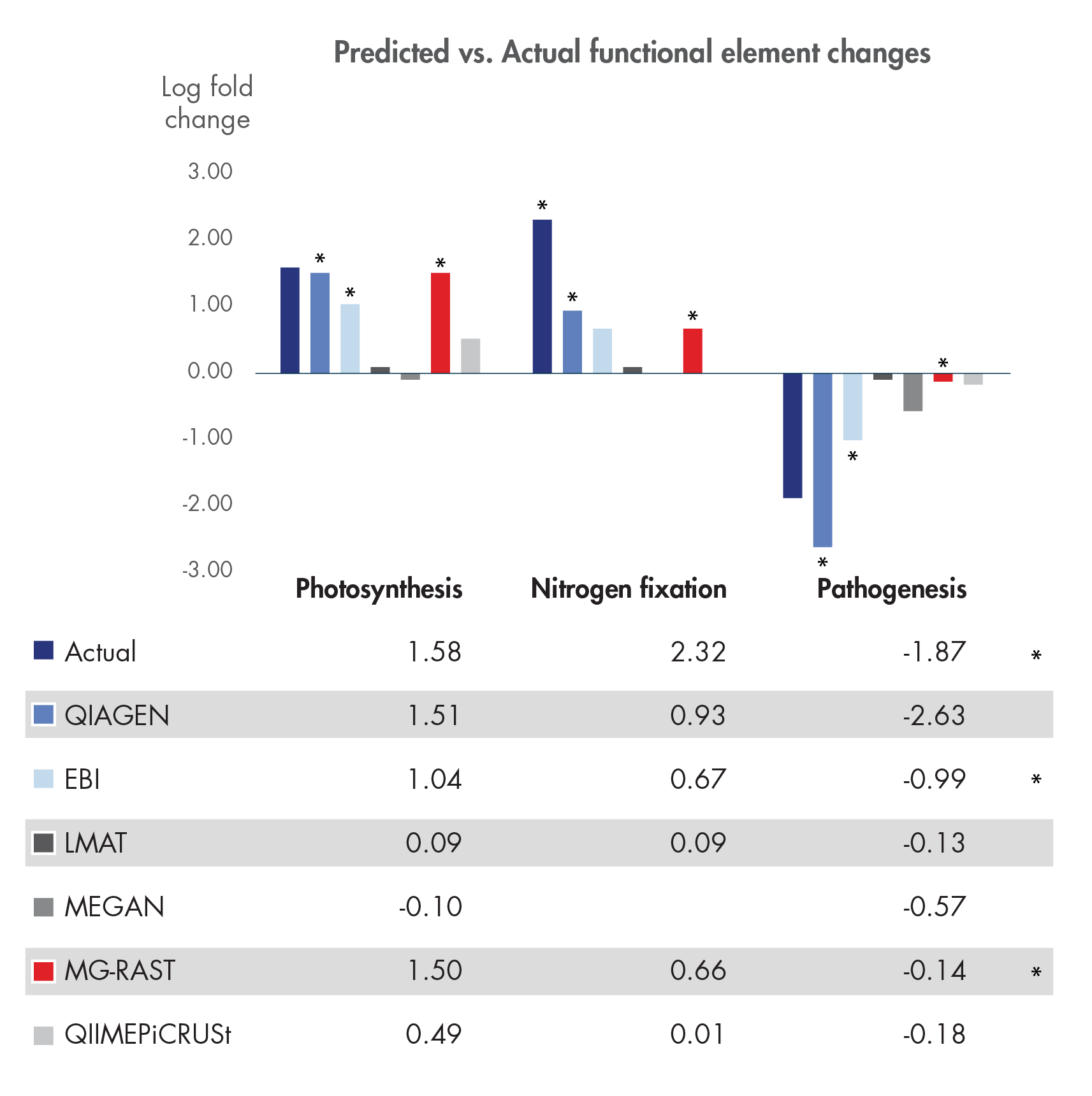
Figure 1. Assigning and tracking gene function in metagenomes with confidence.
Lindgreen et al. published a comprehensive, independent evaluation of 14 different whole metagenome analysis toolkits in Nature Scientific Reports in January 2016. We here compare our solution to the sole five toolkits out of the fourteen that allow functional metagenome analysis using the test data published by Lindgreen et al. Statistical comparison (Edge test performed in CLC Genomics Workbench) of pairwise differential abundance of the individual functional elements predicted in the two test communities detects a statistically significant difference for all of the three functional elements that were analyzed in the paper: photosynthesis, nitrogen fixation and pathogenesis (all p-values < 0.01). Fold-changes predicted using our tools capture the expected overall pattern of functional changes and estimate the actual fold-change with higher precision than any other tool in all three functional roles.
* indicates tools that consistently predicted changes correctly with statistically significance.
Detecting gene function in microbial communities based on metagenomic data is hard. Correctly measuring changes in the functional makeup between different metagenome samples is even harder.
Lindgreen et al. showed that most of the benchmarked open source tools failed to correctly predict such changes at levels that are statistically significant.
With our solution for microbial genomics you can more accurately detect and quantify functional elements in a sample. And the included statistical tools allow you to confidently measure statistically significant changes in function between samples.
Multi-sample comparison is used to detect functional changes between samples and to identify samples with similar or diverging functional genomic elements. Data can be grouped and analyzed in the context of your sample-metadata.
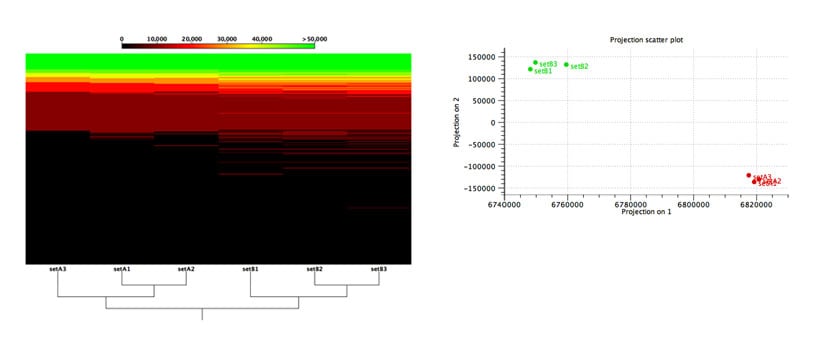
Figure 2: Functional comparison across microbiome samples.
Evaluation of an algorithm’s capabilities in detecting functional changes in metagenomes is notoriously hard because the ground truth is unknown and there exist no gold-standard datasets. To overcome these difficulties, Stinus Lindgreen et al. created six datasets from two synthetic microbial communities for his benchmarks: three (A1, A2 and A3) from the A community and three from the B community. To control the functional content, he created the two communities, A and B, with a selected set of species with known functional capabilities: Cyanobacteria (photosynthesis), Bradyrhizobium (nitrogen fixation) and Rhizobium (nitrogen fixation) were more abundant in community A, while a set of known pathogens where more abundant in community B.
As shown in Figure 2, our tools were able to reliably separate samples from the two different communities based on the relative abundance of their predicted functional content.
Our accurate assignment of gene function depends on a novel metagenome assembler producing higher quality assemblies compared to leading alternatives. Table 1 illustrates how our metagenome assembler compares favorably when it comes to misassemblies, InDels, mismatch errors, and other quality metrics.
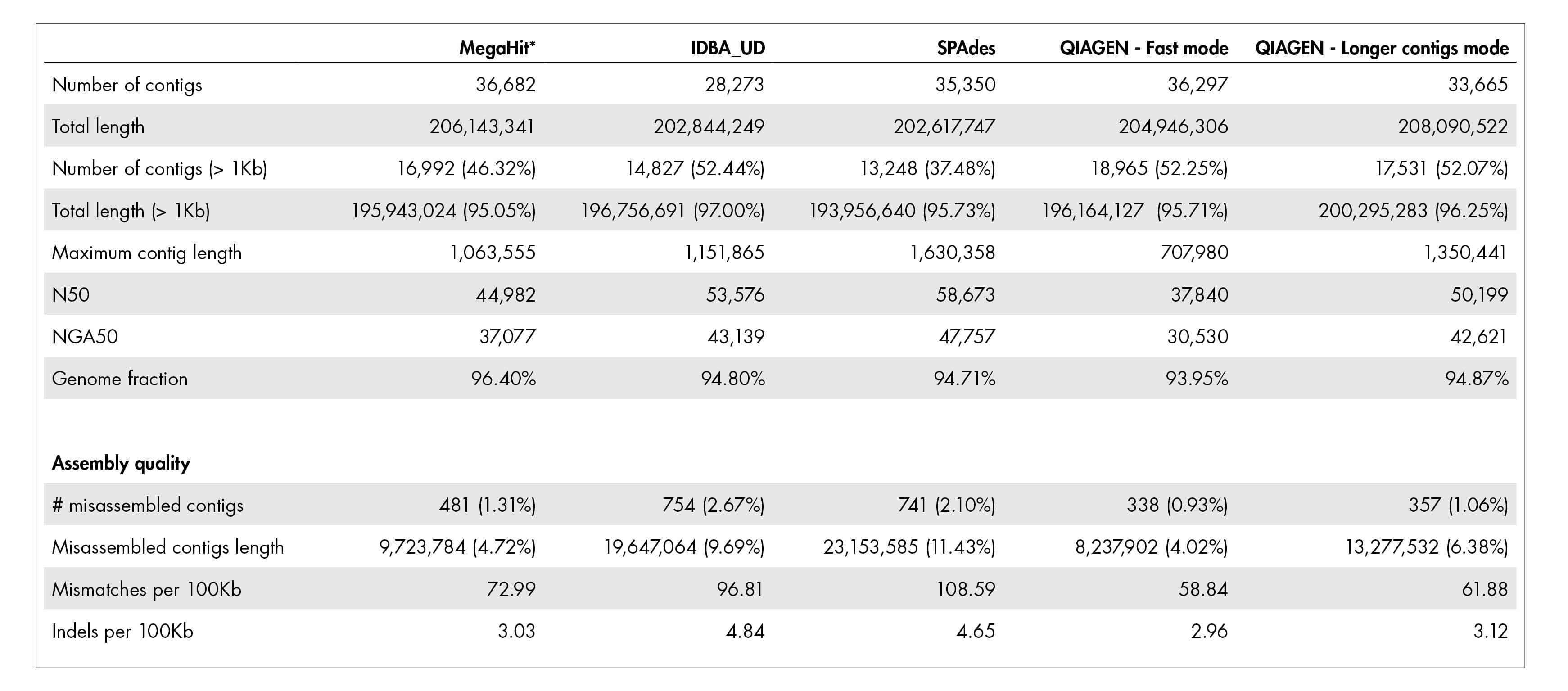
Table 1: Quality of metagenome assembly.
The QIAGEN metagenome assembler delivers superior assembly quality resulting in more accurate annotation of functional genetic content. A dataset published by Shakya et al. 2013 was used for this benchmark. The actual number for “Total length” and “Total length (>1kb)” should be close to 209,845,413 bases.
Run time and compute resource requirements are important when sample volume is high.
We have benchmarked the metagenome assembler included in our microbial genomics solution against leading metagenome assemblers using a dataset by Shakya et al. 2013. Shorter run time and greater compute resource efficiency was consistently demonstrated compared to other leading assemblers.
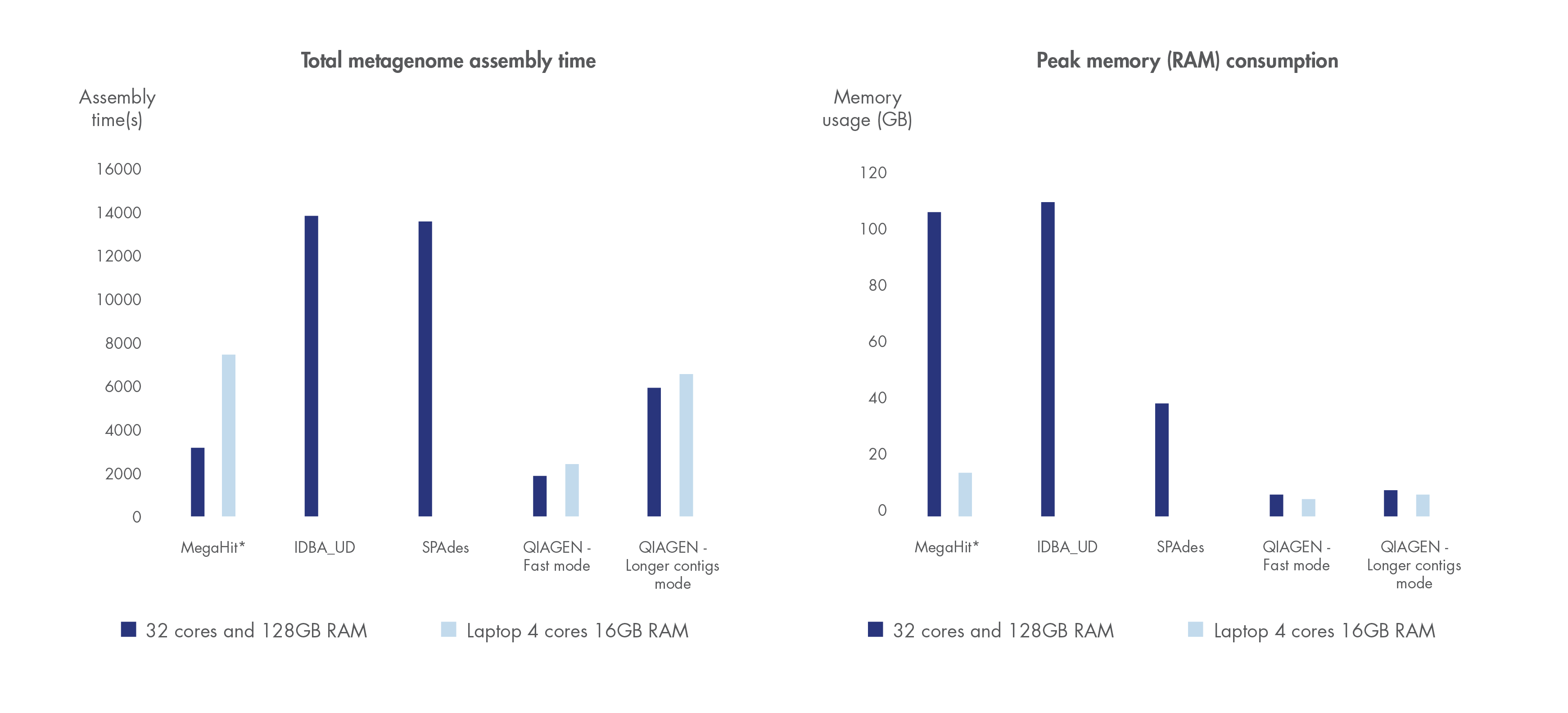
Figure 3. Best in class metagenome assembly.
Accelerated algorithms result in metagenome assembly that outcompetes leading alternatives in run time and compute resource consumption. *Note that MegaHit is able to scale its memory consumption down by sacrificing run time.
Increase walk away time
To increase walk away time, users can use the Workflow feature in CLC Genomics Workbench to combine the analysis steps 2 through 7 listed below into a preconfigured one-click workflow. Workflows are capable of batch processing many samples increasing walk-away time.
Analysis steps in functional metagenomics workflow:
1 → Import of multiple whole metagenome sample read datasets and association of metadata to each sample.
2 → QC and trimming of whole metagenome reads.
3 → De novo assembly of each sample read dataset into high-quality contigs using the new De Novo Assemble Metagenome tool.
4 → Locate coding sequences (CDS) in the resulting contigs using the third-party MetaGeneMark genefinder plugin for the CLC workbenches.
5 → Annotate CDSs with Gene Ontology (GO) terms and Pfam protein families or Best BLAST Hits using one of the two new tools, Annotate CDS with Pfam or Annotate CDS with Best BLAST Hit, respectively.
6 → Map the input reads back to the annotated contigs using the built-in Map Reads to Reference tool in the CLC workbenches.
7 → Build a functional abundance profile of each sample using the Build Functional Profile tool
8 → Merge the functional abundance profiles for all samples into one profile using the Merge Functional Profile tool.
9 → Visualize the individual and merged functional abundance profiles, perform filtering based on abundance, and apply the different options for showing the abundance profiles in the context of metadata.
10→ Perform hierarchical clustering and statistical analysis based on the relative abundance of functional elements in the samples.
Find out more about our microbial genomics solution



