








Ab initio gene finding is a central step in genome analysis, which must account for the biology of the investigated genome(s) in order to perform adequately. Signals are many fold, and include coding potential, hexamer distributions, RNA polymerase-binding and spliceosome-binding sequences, all of which depend on GC content.
The GeneMark family algorithms have been continuously used for genome annotation, starting with the first complete genome (Haemophilus influenza) sequenced in 1995. Currently, an algorithm of the GeneMark family is being used by NCBI as a part of the prokaryotic genome annotation pipeline. Two algorithms, MetaGeneMark and GeneMark-ES, are available as plugins in QIAGEN CLC Genomics Workbench and QIAGEN CLC Genomics Server. MetaGeneMark has proven to deliver accurate gene predictions in metagenomes. GeneMark-ES is an automatic ab initio gene prediction tool for compact eukaryotic genomes. Gene finding in whole genome-sequenced microbial genomes can also be performed using the “Find Prokaryotic Genes” tool of QIAGEN CLC Microbial Genomics Module.
MetaGeneMark
The MetaGeneMark plugin represents a new release of the gene finding algorithm for metagenomic sequences. For each metagenomic contig, MetaGeneMark uses values of the GC content of each ORF in the contig to select sets of gene model parameters (1,2). For a given GC content value, the algorithm uses parameters that vary for archaeal and bacterial domains. This approach ensures that there are no parameters that a user has to select or adjust. The algorithm is fast; it can process 1 GB of metagenomic contigs on a single CPU in less than half an hour.
GeneMark-ES
The GeneMark-ES plugin delivers ab initio predictions of protein-coding genes in eukaryotic genomes (3,4). The GeneMark.hmm algorithm employs a hidden semi-Markov model. The model parameters are determined iteratively using Viterbi training. The most probable parse of a genomic sequence into exons, introns and intergenic regions is thus determined simultaneously with unsupervised training of the model parameters from the genomic sequence, rendering GeneMark-ES a fully automatic tool. GeneMark-ES was shown to produce high gene prediction accuracy for genomes with lengths less than 400 MB. Longer genomes present a challenge due to longer, on average, intergenic regions. The unsupervised training procedure is a computationally expensive task and may take several hours.

Find prokaryotic genes
Ab initio gene finding for microbial genomes can be performed using the “Find Prokaryotic Genes” tool of QIAGEN CLC Microbial Genomics Module. The tool creates a gene prediction model from the input sequence, which estimates GC content, conserved sequences corresponding to ribosomal binding sites, start and stop codon usages, and a statistical model (namely, an Interpolated Markov Model) for estimating the probability of a sequence to be part of a gene compared to the background. The model is then used to predict coding sequences from the input sequence. This tool is inspired by Glimmer3 (5).
Resources
The MetaGeneMark manual and GeneMark-ES manual provide detailed instructions on plugin usage. The use of the algorithms was documented in more than 2000 research publications. The QIAGEN CLC Microbial Genomics Module manual has extensive documentation on the ”Find Prokaryotic Genes” tool and settings and downstream analysis capabilities.
In case RNA-seq data exist, the QIAGEN CLC Genomics Workbench toolbox enables easy verification of ab initio gene predictions, as described in the application note 'Improving structural annotation in complex genomes with QIAGEN CLC Genomics Workbench'.
See also our blog on transcript discovery using QIAGEN CLC Genomics Workbench.
References
Microbial communities contribute more than half of all the cells our bodies are composed of. And not surprisingly, the taxonomic and genetic makeup of microbiomes is closely linked to the health of humans, animals and plants.
Yet especially the functional genetic composition of microbiomes is hard to establish and current metagenomics tools struggle with correctly predicting functional composition or changes in function between microbiome samples [Lindgreen et al. 2015].
What if you could access tools to de novo assemble metagenome data, reliably predict functional elements, and identify statistically significant changes in function between samples? And what if these tools were fully integrated into the industry standard for scientist-friendly NGS data analysis, and came along with a toolbox that has been optimized for microbiologists?
CLC Genomics Workbench, CLC Microbial Genomics Module and the MetaGeneMark plugin deliver superior performance, a fully integrated user experience and come bundled at a competitive price.
Accuracy of results
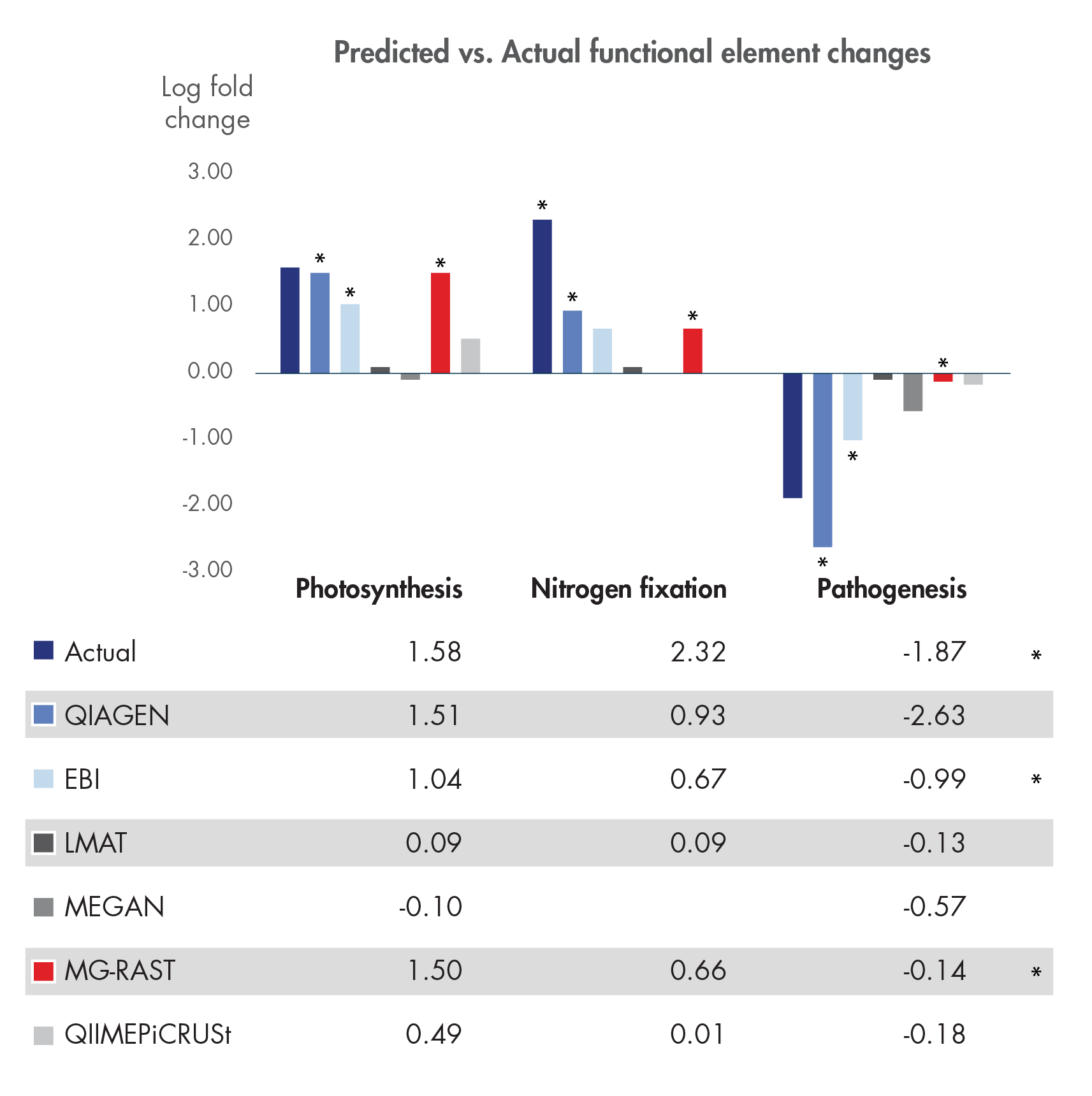
Figure 1. Assigning and tracking gene function in metagenomes with confidence.
Lindgreen et al. published a comprehensive, independent evaluation of 14 different whole metagenome analysis toolkits in Nature Scientific Reports in January 2016. We here compare our solution to the sole five toolkits out of the fourteen that allow functional metagenome analysis using the test data published by Lindgreen et al. Statistical comparison (Edge test performed in CLC Genomics Workbench) of pairwise differential abundance of the individual functional elements predicted in the two test communities detects a statistically significant difference for all of the three functional elements that were analyzed in the paper: photosynthesis, nitrogen fixation and pathogenesis (all p-values < 0.01). Fold-changes predicted using our tools capture the expected overall pattern of functional changes and estimate the actual fold-change with higher precision than any other tool in all three functional roles.
* indicates tools that consistently predicted changes correctly with statistically significance.
Detecting gene function in microbial communities based on metagenomic data is hard. Correctly measuring changes in the functional makeup between different metagenome samples is even harder.
Lindgreen et al. showed that most of the benchmarked open source tools failed to correctly predict such changes at levels that are statistically significant.
With our solution for microbial genomics you can more accurately detect and quantify functional elements in a sample. And the included statistical tools allow you to confidently measure statistically significant changes in function between samples.
Multi-sample comparison is used to detect functional changes between samples and to identify samples with similar or diverging functional genomic elements. Data can be grouped and analyzed in the context of your sample-metadata.
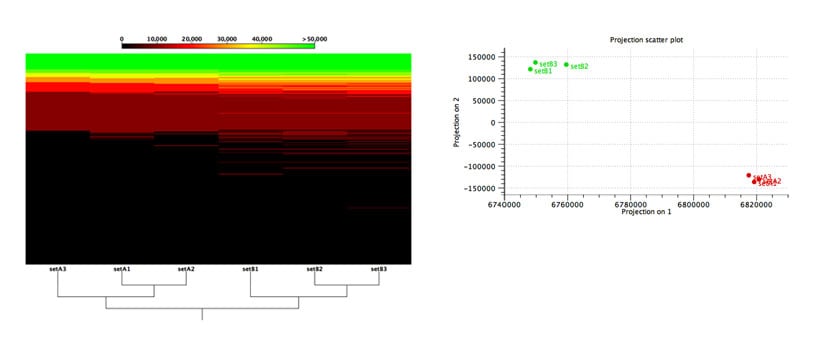
Figure 2: Functional comparison across microbiome samples.
Evaluation of an algorithm’s capabilities in detecting functional changes in metagenomes is notoriously hard because the ground truth is unknown and there exist no gold-standard datasets. To overcome these difficulties, Stinus Lindgreen et al. created six datasets from two synthetic microbial communities for his benchmarks: three (A1, A2 and A3) from the A community and three from the B community. To control the functional content, he created the two communities, A and B, with a selected set of species with known functional capabilities: Cyanobacteria (photosynthesis), Bradyrhizobium (nitrogen fixation) and Rhizobium (nitrogen fixation) were more abundant in community A, while a set of known pathogens where more abundant in community B.
As shown in Figure 2, our tools were able to reliably separate samples from the two different communities based on the relative abundance of their predicted functional content.
Our accurate assignment of gene function depends on a novel metagenome assembler producing higher quality assemblies compared to leading alternatives. Table 1 illustrates how our metagenome assembler compares favorably when it comes to misassemblies, InDels, mismatch errors, and other quality metrics.
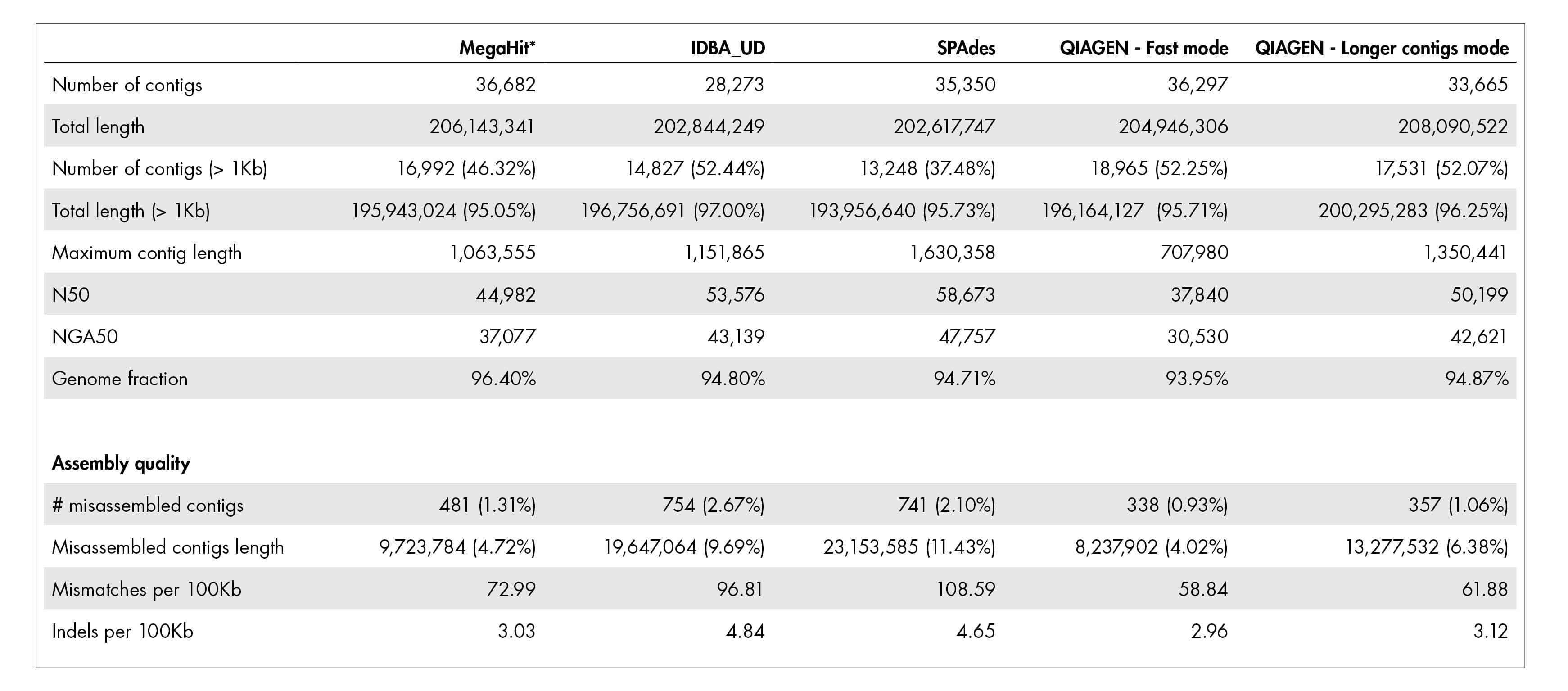
Table 1: Quality of metagenome assembly.
The QIAGEN metagenome assembler delivers superior assembly quality resulting in more accurate annotation of functional genetic content. A dataset published by Shakya et al. 2013 was used for this benchmark. The actual number for “Total length” and “Total length (>1kb)” should be close to 209,845,413 bases.
Run time and compute resource requirements are important when sample volume is high.
We have benchmarked the metagenome assembler included in our microbial genomics solution against leading metagenome assemblers using a dataset by Shakya et al. 2013. Shorter run time and greater compute resource efficiency was consistently demonstrated compared to other leading assemblers.
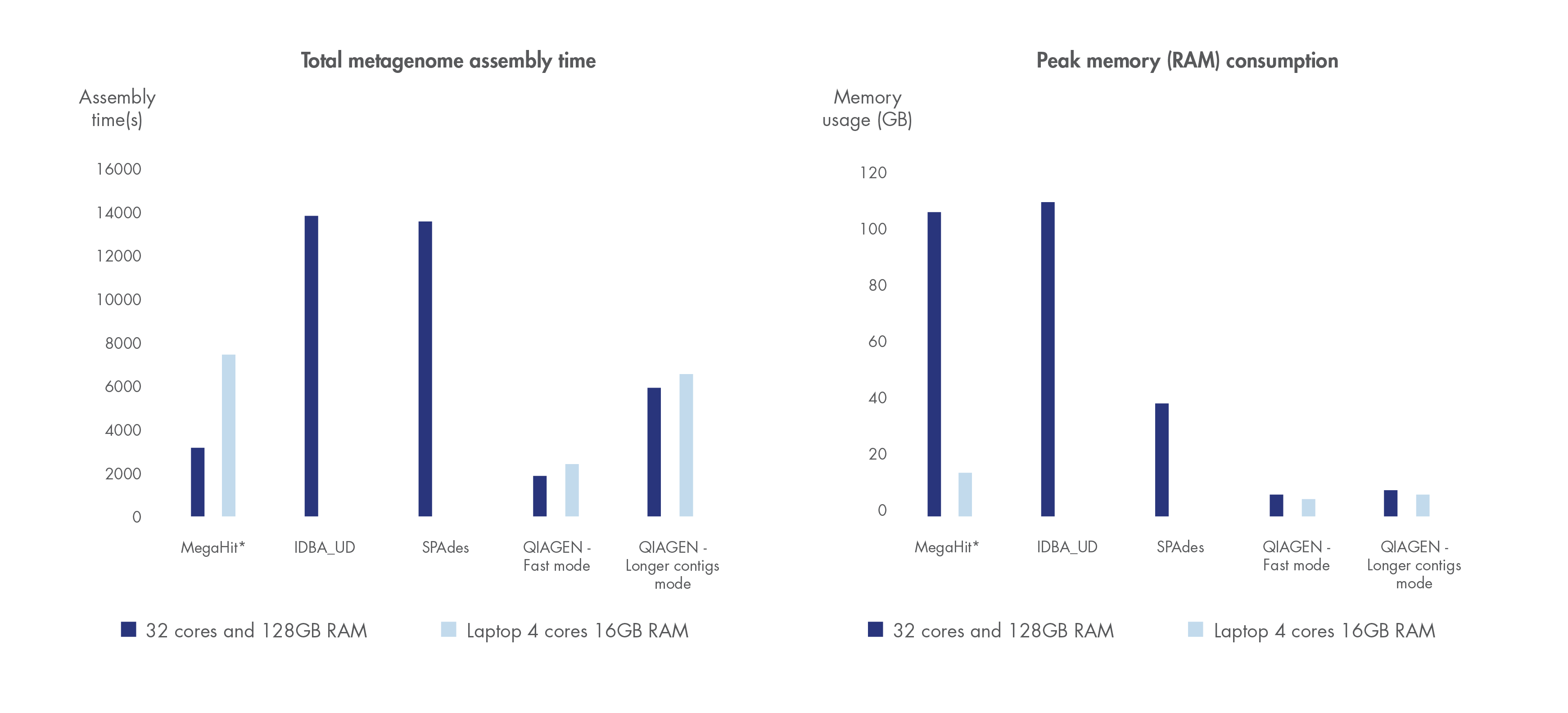
Figure 3. Best in class metagenome assembly.
Accelerated algorithms result in metagenome assembly that outcompetes leading alternatives in run time and compute resource consumption. *Note that MegaHit is able to scale its memory consumption down by sacrificing run time.
Increase walk away time
To increase walk away time, users can use the Workflow feature in CLC Genomics Workbench to combine the analysis steps 2 through 7 listed below into a preconfigured one-click workflow. Workflows are capable of batch processing many samples increasing walk-away time.
Analysis steps in functional metagenomics workflow:
1 → Import of multiple whole metagenome sample read datasets and association of metadata to each sample.
2 → QC and trimming of whole metagenome reads.
3 → De novo assembly of each sample read dataset into high-quality contigs using the new De Novo Assemble Metagenome tool.
4 → Locate coding sequences (CDS) in the resulting contigs using the third-party MetaGeneMark genefinder plugin for the CLC workbenches.
5 → Annotate CDSs with Gene Ontology (GO) terms and Pfam protein families or Best BLAST Hits using one of the two new tools, Annotate CDS with Pfam or Annotate CDS with Best BLAST Hit, respectively.
6 → Map the input reads back to the annotated contigs using the built-in Map Reads to Reference tool in the CLC workbenches.
7 → Build a functional abundance profile of each sample using the Build Functional Profile tool
8 → Merge the functional abundance profiles for all samples into one profile using the Merge Functional Profile tool.
9 → Visualize the individual and merged functional abundance profiles, perform filtering based on abundance, and apply the different options for showing the abundance profiles in the context of metadata.
10→ Perform hierarchical clustering and statistical analysis based on the relative abundance of functional elements in the samples.
Find out more about our microbial genomics solution



